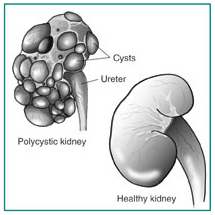
Polycystic kidney disease (PKD) is a genetic disorder characterized by the growth of numerous cysts in the kidneys. The kidneys are two organs, each about the size of a fist, located in the upper part of a person's abdomen, toward the back. The kidneys filter wastes and extra fluid from the blood to form urine. They also regulate amounts of certain vital substances in the body. When cysts form in the kidneys, they are filled with fluid. PKD cysts can profoundly enlarge the kidneys while replacing much of the normal structure, resulting in reduced kidney function and leading to kidney failure.
When PKD causes kidneys to fail-which usually happens after many years-the patient requires dialysis or kidney transplantation. About one-half of people with the most common type of PKD progress to kidney failure, also called end-stage renal disease (ESRD).
PKD can also cause cysts in the liver and problems in other organs, such as blood vessels in the brain and heart. The number of cysts as well as the complications they cause help doctors distinguish PKD from the usually harmless "simple" cysts that often form in the kidneys in later years of life.
In the United States, about 600,000 people have PKD, and cystic disease is the fourth leading cause of kidney failure. Two major inherited forms of PKD exist:
Autosomal dominant PKD is the most common inherited form. Symptoms usually develop between the ages of 30 and 40, but they can begin earlier, even in childhood. About 90 percent of all PKD cases are autosomal dominant PKD.
Autosomal recessive PKD is a rare inherited form. Symptoms of autosomal recessive PKD begin in the earliest months of life, even in the womb.
What is autosomal dominant PKD?
Autosomal dominant PKD is the most common inherited disorder of the kidneys. The phrase "autosomal dominant" means that if one parent has the disease, there is a 50 percent chance that the disease gene will pass to a child. In some cases-perhaps 10 percent-autosomal dominant PKD occurs spontaneously in patients. In these cases, neither of the parents carries a copy of the disease gene.
Many people with autosomal dominant PKD live for several decades without developing symptoms. For this reason, autosomal dominant PKD is often called "adult polycystic kidney disease." Yet, in some cases, cysts may form earlier in life and grow quickly, causing symptoms in childhood.
[ Picture of polycystic kidney, roughly retains the same shape as the healthy kidney. ]
The cysts grow out of nephrons, the tiny filtering units inside the kidneys. The cysts eventually separate from the nephrons and continue to enlarge. The kidneys enlarge along with the cysts-which can number in the thousands-while roughly retaining their kidney shape. In fully developed autosomal dominant PKD, a cyst-filled kidney can weigh as much as 20 to 30 pounds. High blood pressure is common and develops in most patients by age 20 or 30.
What are the symptoms of autosomal dominant PKD?
The most common symptoms are pain in the back and the sides (between the ribs and hips), and headaches. The dull pain can be temporary or persistent, mild or severe.
People with autosomal dominant PKD also can experience the following complications : -
Urinary tract infections, specifically in the kidney cysts
Hematuria (blood in the urine)
Liver and pancreatic cysts
Abnormal heart valves
High blood pressure
Kidney stones;
Aneurysms (bulges in the walls of blood vessels) in the brain
Diverticulosis (small pouches bulge outward through the colon).
How is autosomal dominant PKD diagnosed?
Autosomal dominant PKD is usually diagnosed by kidney imaging studies. The most common form of diagnostic kidney imaging is ultrasound, but more precise studies, such as computerized tomography (CT) scans or magnetic resonance imaging (MRI) are also widely used. In autosomal dominant PKD, the onset of kidney damage and how quickly the disease progresses can vary. Kidney imaging findings can also vary considerably, depending on a patient's age. Younger patients usually have both fewer and smaller cysts. Doctors have therefore developed specific criteria for diagnosing the disease with kidney imaging findings, depending on patient age. For example, the presence of at least two cysts in each kidney by age 30 in a patient with a family history of the disease can confirm the diagnosis of autosomal dominant PKD. If there is any question about the diagnosis, a family history of autosomal dominant PKD and cysts found in other organs make the diagnosis more likely.
In most cases of autosomal dominant PKD, patients have no symptoms and their physical condition appears normal for many years, so the disease can go unnoticed. Physical checkups and blood and urine tests may not lead to early diagnosis. Because of the slow, undetected progression of cyst growth, some people live for many years without knowing they have autosomal dominant PKD.
Once cysts have grown to about one-half inch, however, diagnosis is possible with imaging technology. Ultrasound, which passes sound waves through the body to create a picture of the kidneys, is used most often. Ultrasound imaging does not use any injected dyes or radiation and is safe for all patients, including pregnant women. It can also detect cysts in the kidneys of a fetus, but large cyst growth this early in life is uncommon in autosomal dominant PKD.
More powerful and expensive imaging procedures such as CT scans and MRI also can detect cysts. Recently, MRI has been used to measure kidney and cyst volume and monitor kidney and cyst growth, which may serve as a way to track progression of the disease.
Diagnosis can also be made with a genetic test that detects mutations in the autosomal dominant PKD genes, called PKD1 and PKD2. Although this test can detect the presence of the autosomal dominant PKD mutations before large cysts develop, its usefulness is limited by two factors: detection of a disease gene cannot predict the onset of symptoms or ultimate severity of the disease, and if a disease gene is detected, no specific prevention or cure for the disease exists. However, a young person who knows of a PKD gene mutation may be able to forestall the loss of kidney function through diet and blood pressure control. The genetic test may also be used to determine whether a young member of a PKD family can safely donate a kidney to a family member with the disease. Individuals with a family history of PKD who are of childbearing age might also want to know whether they have the potential of passing a PKD gene to a child. Anyone considering genetic testing should receive counseling to understand all the implications of the test.
How is autosomal dominant PKD treated?
Although a cure for autosomal dominant PKD is not available, treatment can ease symptoms and prolong life.
Pain : - Pain in the area of the kidneys can be caused by cyst infection, bleeding into cysts, kidney stone, or stretching of the fibrous tissue around the kidney with cyst growth. A doctor will first evaluate which of these causes are contributing to the pain to guide treatment. If it is determined to be chronic pain due to cyst expansion, the doctor may initially suggest over-the-counter pain medications, such as aspirin or acetaminophen (Tylenol). Consult your doctor before taking any over-the-counter medication because some may be harmful to the kidneys. For most but not all cases of severe pain due to cyst expansion, surgery to shrink cysts can relieve pain in the back and sides. However, surgery provides only temporary relief and does not slow the disease's progression toward kidney failure.
Headaches : - that are severe or that seem to feel different from other headaches might be caused by aneurysms-blood vessels that balloon out in spots-in the brain. These aneurysms could rupture, which can have severe consequences. Headaches also can be caused by high blood pressure. People with autosomal dominant PKD should see a doctor if they have severe or recurring headaches-even before considering over-the-counter pain medications.
Urinary tract infections : - People with autosomal dominant PKD tend to have frequent urinary tract infections, which can be treated with antibiotics. People with the disease should seek treatment for urinary tract infections immediately because infection can spread from the urinary tract to the cysts in the kidneys. Cyst infections are difficult to treat because many antibiotics do not penetrate the cysts.
High blood pressure : - Keeping blood pressure under control can slow the effects of autosomal dominant PKD. Lifestyle changes and various medications can lower high blood pressure. Patients should ask their doctors about such treatments. Sometimes proper diet and exercise are enough to keep blood pressure controlled.
End-stage renal disease : - After many years, PKD can cause the kidneys to fail. Because kidneys are essential for life, people with ESRD must seek one of two options for replacing kidney functions: dialysis or transplantation. In hemodialysis, blood is circulated into an external filter, where it is cleaned before re-entering the body; in peritoneal dialysis, a fluid is introduced into the abdomen, where it absorbs wastes and is then removed. Transplantation of healthy kidneys into ESRD patients has become a common and successful procedure. Healthy-non-PKD-kidneys transplanted into PKD patients do not develop cysts.
What is autosomal recessive PKD?
Autosomal recessive PKD is caused by a mutation in the autosomal recessive PKD gene, called PKHD1. Other genes for the disease might exist but have not yet been discovered by scientists. We all carry two copies of every gene. Parents who do not have PKD can have a child with the disease if both parents carry one copy of the abnormal gene and both pass that gene copy to their baby. The chance of the child having autosomal recessive PKD when both parents carry the abnormal gene is 25 percent. If only one parent carries the abnormal gene, the baby cannot get autosomal recessive PKD but could ultimately pass the abnormal gene to his or her children.
The signs of autosomal recessive PKD frequently begin before birth, so it is often called "infantile PKD." Children born with autosomal recessive PKD often, but not always, develop kidney failure before reaching adulthood. Severity of the disease varies. Babies with the worst cases die hours or days after birth due to respiratory difficulties or respiratory failure.
Some people with autosomal recessive PKD do not develop symptoms until later in childhood or even adulthood. Liver scarring occurs in all patients with autosomal recessive PKD and tends to become more of a medical concern with increasing age.
What are the symptoms of autosomal recessive PKD?
Children with autosomal recessive PKD experience high blood pressure, urinary tract infections, and frequent urination. The disease usually affects the liver and spleen, resulting in low blood cell counts, varicose veins, and hemorrhoids. Because kidney function is crucial for early physical development, children with autosomal recessive PKD and decreased kidney function are usually smaller than average size. Recent studies suggest that growth problems may be a primary feature of autosomal recessive PKD.
How is autosomal recessive PKD diagnosed?
Ultrasound imaging of the fetus or newborn reveals enlarged kidneys with an abnormal appearance, but large cysts such as those in autosomal dominant PKD are rarely seen. Because autosomal recessive PKD tends to scar the liver, ultrasound imaging of the liver also aids in diagnosis.
How is autosomal recessive PKD treated?
Medicines can control high blood pressure in autosomal recessive PKD, and antibiotics can control urinary tract infections. Eating increased amounts of nutritious food improves growth in children with autosomal recessive PKD. In some cases, growth hormones are used. In response to kidney failure, autosomal recessive PKD patients must receive dialysis or transplantation. If serious liver disease develops, some people can undergo combined liver and kidney transplantation.
What are genetic diseases?
Genes are segments of DNA, the long molecules that reside in each of a person's cells. The genes, through complex processes, build proteins for growth and maintenance of the body. At conception, DNA-or genes-from both parents are passed to the child.
A genetic disease occurs when one or both parents pass abnormal genes to a child at conception. If receiving an abnormal gene from just one parent is enough to produce a disease in the child, the disease is said to have dominant inheritance. If receiving abnormal genes from both parents is needed to produce disease in the child, the disease is said to be recessive. A genetic disease can also occur through a spontaneous mutation.
The chance of acquiring a dominant disease is higher than the chance of acquiring a recessive disease. A child who receives only one gene copy for a recessive disease at conception will not develop the genetic disease-such as autosomal recessive PKD-but could pass the gene to the following generation.
Hope through research
Scientists have begun to identify the processes that trigger formation of PKD cysts. Advances in the field of genetics have increased our understanding of the abnormal genes responsible for autosomal dominant and autosomal recessive PKD. Scientists have located two genes associated with autosomal dominant PKD. The first was located in 1985 on chromosome 16 and labeled PKD1. PKD2 was localized to chromosome 4 in 1993. Within 3 years, scientists had isolated the proteins these two genes produce-polycystin-1 and polycystin-2.
When both the PKD1 and PKD2 genes are normal, the proteins they produce work together to foster normal kidney development and inhibit cyst formation. A mutation in either of the genes can lead to cyst formation, but evidence suggests that disease development also requires other factors, in addition to the mutation in one of the PKD genes.
Genetic analyses of most families with PKD confirm mutations in either the PKD1 or PKD2 gene. In about 10 to 15 percent of cases, however, families with autosomal dominant PKD do not show obvious abnormalities or mutations in the PKD1 and PKD2 genes, using current testing methods.
Researchers have also recently identified the autosomal recessive PKD gene, called PKHD1, on chromosome 6. Genetic testing for autosomal recessive PKD to detect mutations in PKHD1 is now offered by a limited number of molecular genetic diagnostics laboratories in the United States.
Researchers have bred rodents with a genetic disease that parallels both inherited forms of human PKD. Studying these mice will lead to greater understanding of the genetic and nongenetic mechanisms involved in cyst formation. In recent years, researchers have discovered several compounds that appear to inhibit cyst formation in mice with the PKD gene. Some of these compounds are in clinical testing in humans. Scientists hope further testing will lead to safe and effective treatments for humans with the disease.
Recent clinical studies of autosomal dominant PKD are exploring new imaging methods for tracking progression of cystic kidney disease. These methods, using MRI, are helping scientists design better clinical trials for new treatments of autosomal dominant PKD.
Polycystic Kidney Disease At A Glance
The two forms of polycystic kidney disease (PKD) are
autosomal dominant PKD, a form that usually causes symptoms in adulthood
autosomal recessive PKD, a rare form that usually causes symptoms in infancy and early childhood
The symptoms and signs of PKD include
pain in the back and lower sides
headaches
urinary tract infections
blood in the urine
cysts in the kidneys and other organs
Diagnosis of PKD is obtained by
ultrasound imaging of kidney cysts
ultrasound imaging of cysts in other organs
family medical history, including genetic testing
PKD has no cure. Treatments include
medicine to control high blood pressure
medicine and surgery to reduce pain
antibiotics to resolve infections
dialysis to replace functions of failed kidneys kidney transplantation
The list of of Nephrology Hospitals in India is as follows : -
For more information, medical assessment and medical quote
send your detailed medical history and medical reports
as email attachment to
Email : - info@wecareindia.com
Call: +91 9029304141 (10 am. To 8 pm. IST)
(Only for international patients seeking treatment in India)
For a detailed evaluation send patient’s medical reports / X rays / doctors notes to info@wecareindia.com
Patient Storys
Successful heart surgery at We Care India partner hospital allows Robert Clarke to live a normal life despite a rare genetic disorder We Care india helped Robert find best super specialised surgeon for his rare conditions.
Polycystic Kidney Disorder Treatment India Offers info on Polycystic Kidney Surgery India, Cost Polycystic Kidney Surgery India, Polycystic Kidney Treatment India, Cost Polycystic Kidney Surgery India, Polycystic Kidney Surgery Hospital India, Polycystic Kidney Treatment India, Low Cost Polycystic Kidney Treatment Delhi India, Polycystic Kidney Treatment Hospital Delhi India, Polycystic Kidney Treatment Mumbai Hospital India, Affordable Polycystic Kidney Hospital India, Low Cost Polycystic Kidney Disorder Treatment India Info Provided By Indiahopsitaltour.Com